柳达:40 年辉煌史,生物药的开发有无范式?
2023-02-19 16:05:58
·
生物制品圈
·
柳达
导读上周,我们通过知名投资专家柳达介绍了化学药范式,本周我们介绍生物药范式。从20世纪80年代到目前,40年来的生物制药历史十分恢弘。此章节中,柳达分析了传统生物制药与具有突破性创新的生物制药企业案例
导读
上周,我们通过知名投资专家柳达介绍了化学药范式,本周我们介绍生物药范式。从20世纪80年代到目前,40年来的生物制药历史十分恢弘。此章节中,柳达分析了传统生物制药与具有突破性创新的生物制药企业案例。他表示,那些具有里程碑意义的药企或药品,一般都不是被战略规划出来的结果。
正如罗氏制药的CEO Severin Schwan所说,“科学的成功不能被规划,但我们可以创造条件,使之得以实现;我们需要对新想法持开放态度,勇于冒险,并偶尔挑战大众普遍持有的观点;我们的研究人员需要自由地研究他们的想法,给他们充足的时间和持久努力的支援”。
正是制药公司里的科学家、研究人员披荆斩棘,于是一个个伟大的生物药就出现了。
从20世纪80年代到目前,40年的生物制药历史。之所以称为“制药”,是因为同化学药相比,生物制药的制造流程、工艺更加重要,直接影响成本及品质控制。依据创新属性,生物制药也可分为生物创新药(Bio-novel)和生物类似药(Bio-similar)。我们采用Dr. Rick Ng的Drugs,From Discovery to Approval的分类方法:1、以胰岛素为代表的替代性蛋白药物;2、以单克隆抗体(简称“单抗”)为代表的治疗性蛋白药物;以及3、各类疫苗产品。在这里,主要介绍替代性蛋白药物及治疗性蛋白药物,由于疫苗具有品类特殊性及目前因Covid-19被广泛关注,我们将有专题重点呈现。本章节比较详细地记录了行业公认的领导者:美国基因泰克(Genentech)及美国安进(Amgen)成长的艰辛及取得的巨大成功。同时,本章也描述其他几个传统的制药行业领导者如何涉足生物制药领域,包括早期的美国礼来(Eli Lilly)、丹麦诺和诺德(Novo Nordisk),及后来的美国辉瑞(Pfizer)、默沙东(Merck)、雅培(Abbott Laboratories)等等。
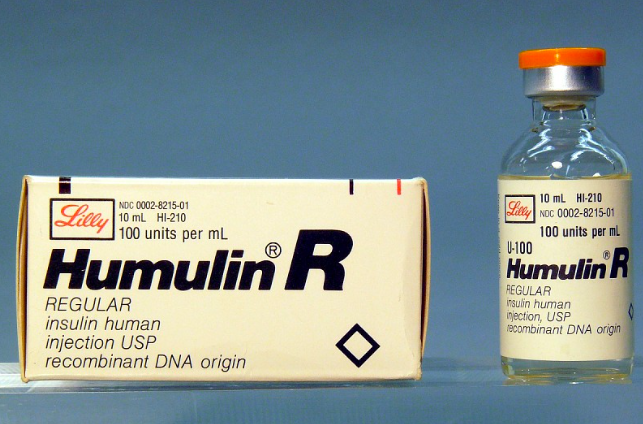
1982年,美国基因泰克(Genentech)和礼来公司(Eli Lilly)共同推出的重组人胰岛素(Humulin)被FDA批准上市,被认为是人类历史上第一个真正意义上的生物制药,这个里程碑开启了生物制药的历史。重组DNA技术从三个方面推进了生物制药的布展:第一,从源头上解决了之前只能以天然材料为原材料而导致的产率低、选择少的问题;第二,为避免使用难以处理或者采集方式危险的原料,提供了可能性;第三,也是最重要的一点,拓展了生产改造蛋白的空间。重组DNA技术的出现真正使制药行业发生“范式转移”,引领这个领域从“化学药时代”进入“生物制药时代”。历史较为悠久的蛋白替代药物相比于治疗性蛋白药率先进入辉煌时期。人类有些疾病是由于人体难以产生某些必须的蛋白,而且缺乏这些蛋白导致一系列健康问题。蛋白替代药就是为患者补充缺乏的蛋白或者天然蛋白的类似物,从而达到治疗疾病的效果。这其中的代表产品有胰岛素、生长激素、凝血因子、白介素等。从上世纪80年代之后近30年间,大量此类产品问世,尤其是活性成分相同的各类改良药,例如:原有药物的改良氨基酸序列药物、新配方及不同用药途径的改良药,和生物类似药。蛋白替代药物的发展从1982年起步。1986年,世界上首个单抗药物Orthoclone OKT3被批准上市,适应症为治疗器官移植后出现的排斥反应,以此拉开了抗体类药物进入现代医学的序幕。自20世纪末期开始到2015年间,每一个五年间隔中被美国FDA和欧盟EMA批准的抗体类药物的数目较为稳定。从2015到2020年,被欧美批准进入市场的抗体类药物的数目与前期同时间间隔相比呈爆发增长状态。在所有被首次批准的生物制药中,抗体类药物超过一半,而在2010-2014年,这个比例只有27%。抗体类药物一枝独秀,以2019年为例。全球最畅销的10款药物中,有8款为抗体类药。1、经典案例之一:胰岛素,礼来制药、诺和诺德、赛诺菲1982年,重组人胰岛素上市。早在20世纪20年代初,胰岛素与糖尿病的关联被发现后不久,从自然动物原料中提取的胰岛素就开始成为治疗糖尿病的药品。1923年,礼来制药推出了美国第一个胰岛素产品。在欧洲,丹麦Nordisk Insulin Laboratory开始大量生产胰岛素,并且出口到丹麦附近的欧洲各国。同源的丹麦公司Novo Therapeutisk也因胰岛素技术迅速发展。从自然原料中提取的动物胰岛素被称为第一代胰岛素。人们很快就意识到自然胰岛素作用时间太短,使用并不方便。而且,从自然原料中,提取有效蛋白成分作为药物有产量低、安全性较差、对于部分患者会引起过敏反应等缺点。常用的第一代胰岛素注射制剂为猪胰岛素,因为其与人胰岛素有几个氨基酸的差别,可能会导致患者免疫反应,使得注射部位皮下脂肪萎缩或者增生。为了延长胰岛素的作用时间,以及减小可能发生的免疫反应,在之后40年,各个公司包括Nordisk、Novo、礼来,都各自突破技术难点,研发出长效胰岛素。例如:Nordisk研发出的精蛋白胰岛素,以及Novo推出的含锌胰岛素。
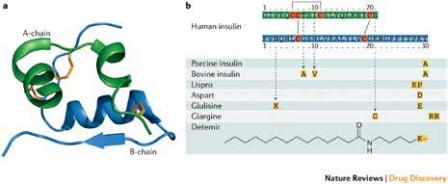
70年代中期,重组DNA技术的发现引发现代医学突破性进步。基因泰克是第一个采用此技术的药物研发公司,成功以大肠杆菌生产出高纯度的合成人胰岛素。其结构和人体自身分泌的胰岛素一样,相比动物胰岛素有较少的过敏反应,促成了重组人胰岛素Humulin的上市。基因泰克由风险投资家Robert A. Swanson和重组DNA技术的先驱者Herbert Boyer博士于1976年创立。这类以重组DNA技术生产出的人胰岛素被称为第二代胰岛素。礼来公司原本就是生产销售第一代胰岛素的领导者,它以独特的眼光比竞争对手抢先一步抓住了第二代胰岛素的机遇,并且与基因泰克合作,主导Humulin的入市审核过程。Humulin也被授权给礼来公司制造,使得它再一次坐稳胰岛素市场。在重组DNA技术的推动下,胰岛素生产成本大幅下降,第二代胰岛素因此逐渐替代第一代胰岛素。1989年,在当时激烈的市场竞争环境下,同在丹麦的Novo公司与Nordisk公司摒弃前嫌,宣布合并。自此,又一胰岛素行业领导者诺和诺德(Novo Nordisk)诞生。从20世纪90年代开始,人们对胰岛素的结构与性质有了更深的了解。随着技术的发展,改造天然人胰岛素结构以调整药物性质成为可能,例如:在胰岛素的肽链上进行修饰,用重组DNA技术改变天然人胰岛素肽链上某些部位的氨基酸序列,改变胰岛素聚体强度等。这种经过改良成为更优秀的胰岛素类似物被称为第三代胰岛素,相比第二代胰岛素,可以在起效时间、峰值时间及作用持续时间上,更好地模拟人体胰岛素的分泌模式。现在市场上畅销的胰岛素产品有不同种类,包括超长效胰岛素、速效胰岛素、超短效胰岛素、甚至吸入式胰岛素等。图表2:人胰岛素及各胰岛素类似物的氨基酸序列和结构第三代胰岛素的发展过程中,值得了解的是吸入式胰岛素的曲折发展。2006年,辉瑞制药获得FDA批准全球第一个吸入性胰岛素产品Exubera,被认为是生物制药史上的重大突破,因为其他胰岛素生物制药的给药途径都是注射。然而,其销售极为惨淡,辉瑞制药在2007年宣布Exubera退市。Exubera的失败也使诺和诺德与礼来制药中断相类似的吸入式胰岛素研究项目。然而,这些挫折并没有影响美国Mann Kind公司的决策,其吸入性胰岛素产品Afrezza在2014年获批。Highlands Pharmaceuticals公司的类似产品Dypreza在2013年及2016年分别在欧洲及美国获批。2018年,产品因长期通过肺部吸收胰岛素的安全隐患问题被召回。美国Nektar Therapeutics是Exubera的最初发明者。辉瑞制药在1995年就与Nektar签署关于这项产品的授权与开发协定。辉瑞制药的Lipitor在2006年的销售额为130亿美元,占了辉瑞全年收入的27%,其在美国市场专利保护于2011年到期前,辉瑞制药急于寻找一款“重磅炸弹”产品来替代Lipitor。辉瑞制药预测Exubera会是一个“超级重磅炸弹”,并且在2006年花费13亿美元买断了法国赛诺菲—安万特(Sanofi-Aventis)的份额,使得辉瑞拥有其全球的生产销售权利。然后,辉瑞制药耗费了11年时间进行研发的产品,上市1年就失败了。这款药物失败的原因引发了各方的讨论,原因之一是Exubera吸入器的设计。这种吸入器尺寸大小与标准网球罐相若,远大于市场上现有普通吸入器,便携性较差。另一个原因是辉瑞制药松散的市场销售管理。辉瑞在Exubera市场化的过程中准备相当不足。即便如此,吸入性胰岛素产品并没有完全失败,MannKind公司的Afrezza在市场上依然有销售。除了吸入式胰岛素,口服胰岛素也为新型研发热点,可解决目前注射性胰岛素的病人依从性问题。该领域较领先的公司为以色列的Oramed Pharmaceuticals,其研发的重组人胰岛素肠溶胶囊(ORMD-0801)目前已同时进入美国及中国临床III期。在胰岛素领域,全球基本形成三足鼎立的局面:礼来制药,赛诺菲和诺和诺德。赛诺菲凭藉一款甘精胰岛素Lantus奠定市场地位,但是近期它们决定退出对胰岛素的研发。2、经典案例之二:生长激素(Human Growth Hormone,hGH),基因泰克、诺和诺德人胰岛素并不是基因泰克公司研发的第一个产品。当时,基因泰克希望通过完成比较艰巨的项目来证明公司的能力,并且拓展公司在药物生产、申报和销售方面的能力,促使基因泰克成为一个独立完整的制药公司。人胰岛素一共有51个氨基酸,而人生长激素有191个氨基酸。生长激素的生产技术难度是远大于胰岛素的技术。早期的基因泰克以科学家为主导,雄心勃勃地选择开发生长激素作为优先顺序更高的研发产品,即便在当时的估算下生长激素市场远没有胰岛素大。生长激素是由人脑干中的垂体分泌,垂体直径约10毫米,大约蚕豆大小。生长激素对人的生长、代谢和发育都起到非常重要的作用,例如:可以调节儿童身高增长、刺激脂肪分解、促进蛋白合成,甚至调节糖类代谢和电解质平衡。生长激素不同于胰岛素,动物自然原料中提取的动物生长激素对人类并不起作用,所以只有人类生长激素可以作为药物使用。在重组生长激素诞生之前,生长激素蛋白药只能从人类垂体中提取。这个来源非常少,大致是遗体捐献或者有需要切除垂体治疗其他疾病的患者捐献。垂体本身又很小,能提取到的生长激素更是有限。以至于美国甚至在1960年成立了国家垂体机构(National Pituitary Agency)专门管辖捐献垂体的合理分配。基因泰克的项目是希望解决生长激素供应的问题。
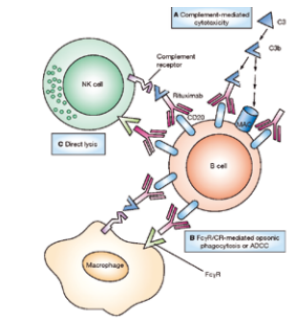
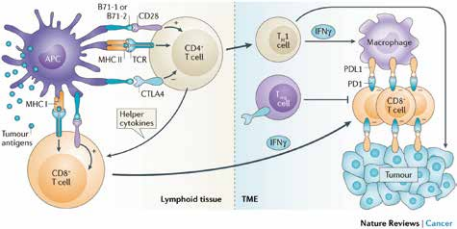
生长激素项目从开始到最终成功,也是生物制药进步的缩影。首先,在商业方面,基因泰克首创“首付款+达成里程碑付款+销售提成”的项目转让模式。在研发生长激素的过程中,基因泰克遭遇现金流的问题,便将研发中的生长激素项目的未来权益转让给瑞典Kabi公司。Kabi一直在欧洲销售,提取生长激素,因此对这个项目产生兴趣。然而,这个项目的研发属相对早期阶段,为了规避研发失败所带来的交易损失,基因泰克就提出了将合作支付资金平摊到整个产品研发到上市的过程中的转让模式。双方达成协定后,Kabi先付给基因泰克一部分钱,然后按照双方约定的阶段目标,基因泰克每完成一个目标,Kabi就支付一笔费用,直到项目成功,Kabi将拥有这款产品在欧洲的权益。1978年,基因泰克与Kabi签署协定,成为历史上第一例生物技术公司与制药公司的合作。这种首创的项目转让模式也成为后来公司间项目合作的常规做法。其次,在技术方面,基因泰克首创用细菌表达纯的、有生物活性的生长激素,这是用细胞系第一次表达全蛋白。1979年,基因泰克在《自然》杂志(Nature)上发表用大肠杆菌表达人生长激素的论文。基因泰克突破了许多技术难点,为以后的发展项目奠定坚实的基础。第三,在政府监管方面,基因泰克的经历也体现药物注册审批上的挑战与难度。基因泰克生产出的第一代生长激素Protropin,与天然提取生长激素相比,基因泰克的产品并未展现出更好的疗效,却隐含了免疫排斥的风险,因此迟迟未有得到FDA批准。基因泰克从1982年底开始药物申报,直到1985年,人们发现自然提取的生长激素中有可能带有朊病毒(Prion),因此在美国被禁止使用。同年,FDA批准了基因泰克的重组人生长激素作为孤儿药治疗生长激素缺陷型儿童适应症,才使得生长激素类产品迎来了曙光。几大制药公司在生长激素类产品上进行改良和市场竞争,例如:礼来制药在1987年推出不带多馀氨基酸的重组人生长激素Humatrope,被FDA批准为孤儿药治疗生长激素缺陷型儿童适应症。这一举措将基因泰克开发的同样不带多馀氨基酸的重组人生长激素Nutropin排除在这个适应症之外。基因泰克只能另辟蹊径,寻求生长激素的其他适应症,例如:儿童慢性肾功能不全导致的生长迟缓症、特纳综合症(Turner Syndrome)、成人生长激素缺乏症等。生长激素的适应症不断被拓展的同时,也流行于各种标签外使用(Off-label use),例如:在防止衰老领域(Anti-aging)。诺和诺德公司在闯入美国市场后,其产品Norditropin迅速抢占大量市场份额。后来者居上,反而超越基因泰克。究其原因,是诺和诺德不只是提供一款药品,而是做成一个产品加服务的组合,为患者解决使用过程中的问题。诺和诺德的人生长激素注射笔对于儿童,针头细且注射痛苦小,更加容易被接受;而对家长,使用方便、计量准确,甚至药品在几周内不需冷藏。另外,为了配合美国的医疗保险报销体系,诺和诺德专门派团队帮助选择使用这款产品的家庭处理保险事务。良好的用户体验使得这款产品大受欢迎。3、经典案例之三:促红血球生成素(Erythropoietin,EPO),因专利纷争而发展的生物制药,安进、强生制药促红血球生成素由肾脏分泌,是一种促进人体生成血红细胞的糖蛋白激素。作为药物,以重组DNA技术细胞合成的促红血球生成素类药物被统称为红血球生成刺激剂(Erythropoiesis stimulating agent,ESA),主要用于治疗人类贫血症。人类历史上第一个ESA药物是由安进公司完成,随之而来的是安进公司与强生公司(JNJ.US)在专利纠纷上的问题。不同于胰岛素与生长激素类药物,20世纪70年代EPO及其功能被发现后,人类一直没有找到可以从自然原料中提取足量EPO作为药物的方法。安进公司科学家林福坤及其团队在简陋的试验条件下,从150万个人的基因碎片中找到EPO基因片段,为人类历史上又一经典药物Epogen的诞生做出卓越贡献。1985年,安进公司的EPO候选药物第一次进入临床试验,适应症为与肾病相关的贫血症。但是,安进公司却在此时遭遇了严重的资金困难。于是,安进公司找到强生制药合作,将此候选药物除肾病贫血以外的适应症在美国和欧洲的市场销售权卖给强生制药。这个EPO候选药物在临床试验中取得了令人瞩目的试验结果。1989年6月,美国FDA批准Epogen的上市申请,用于治疗由慢性肾病引起的贫血症,是历史上第一个被批准的红血球生成刺激剂,也是安进公司被批准的第一款药物。EPO一上市就成为明星,并被评为年度最创新药物。后来,强生制药以所签订的协议为基础,将安进公司告上法庭。在1989年3月,法院就判决这两家公司必须提交联合申请,以交叉授权的方式将产品推出市场。两家公司的专利官司持续十年之久。在1998年,法院判定安进公司因违反Epogen销售协定向强生赔款2亿美元,而强生则需要赔偿安进公司在长期争议中损失的1亿美元。这场专利官司,无论对哪一方都是时间与资源的损失。安进公司所想的出路便是研发改良药品,在原有药品分子的基础上加上化学修饰,使其变得更为长效。2001年,安进公司研发的这款改良版的EPO药物Aranesp获得美国FDA批准,是一个全新的分子,不受当年专利协定的限制。胰岛素类、生长激素类、EPO类产品,作为蛋白替代药物的重要代表,反映了这类药物的发展史。从2015年起,被美国FDA和欧盟EMA批准的蛋白替代药物种类与数目都有减少的趋势,反映这类市场逐渐饱和。可以预计,虽然蛋白替代药物依旧会作为带来人类健康福祉的重要药物活跃在市场上,各大型制药公司也会运用各自几十年的知识技术积累不断改良现有药物,此类药物出现重大突破的可能性变低。尤其是小型初创公司,在没有重大技术突破的情况下,依靠蛋白替代药物脱颖而出的概率不大。抗体类药物取得如此巨大的成功不是偶然。与传统化学药相比,抗体药物与蛋白靶点结合特异性高,结合能力强,脱靶率低,可以避免使用化学药的代谢过程或者产物有肝肾毒性的问题,进而疗效更好、副作用更小。相比于蛋白替代药物,抗体类药物分子量更大、结构更加复杂。这类药物经常有复杂的翻译后修饰结构,尤其是糖基化结构。抗体的糖基化结构极大影响抗体药的药效与半衰期。但是,经常用于生产蛋白替代药物的细胞系,例如:大肠杆菌,就无法生产出有如此复杂结构且带有适当糖基的抗体药。直到1975年,科学家Kohler与Milstein第一次创造性生产单克隆抗体的杂交瘤细胞系,才使得生产和研究单克隆抗体成为可能,以此奠定了抗体类药物发展基础。如何筛选研发的抗体药物蛋白分子使其成为有效安全的治疗药物是研发的重要因素,而是否可以将蛋白分子以合适的成本大量生产则是另一个相辅相成的关注事项。在抗体药物发展的早期阶段,找到适合生产抗体类蛋白药物的体系就成了行业中亟待解决的技术难点。为了满足大规模工业生产的要求,全球制药研发机构以及科学家们积极探索各种可能的生产方法,以至于在2006-2015年间,被批准的抗体药生产体系呈现百家争鸣的态势。2015年后,哺乳动物细胞系逐渐占据主导地位,而抗体类药物的生产过程也趋于成熟。这为抗体类药物进入高速发展奠定了基础。基因泰克是全球生物科技的先驱者,在抗体药的发展中做出了卓越的贡献,成功开发出多款经典抗体药物,长期占据全球年度销售榜前十,其中最出名的就是“抗癌三杰”:美罗华、赫赛汀和阿瓦斯汀。1990年,罗氏制药出资21亿美元收购了基因泰克60%的股份,解决了基因泰克当时因研发投入过大出现严重现金周转不足的燃眉之急。随后的20年间,基因泰克持续推出“重磅炸弹”药物,让罗氏制药意识到基因泰克的非凡价值。2009年,罗氏制药耗资468亿美元全额收购了基因泰克。1、经典案例之一:治疗液体肿瘤,抗CD20抗体药物,Rituxan1997年,美罗华(Rituxan)获得美国FDA批准,成为第一个上市的抗癌单克隆抗体药物。美罗华的有效成分是利妥昔单抗(Rituximab),是抗CD20人鼠嵌合单克隆抗体。CD20蛋白是一种广泛分布在恶性B细胞上的蛋白,在1980年被丹娜—法伯癌症研究所(Dana Farber Cancer Institute)的研究员Lee Nadler发现。单克隆抗体与CD20结合可以触发细胞凋亡,达到杀死癌细胞的作用。自CD20被发现之后,以其为靶点的单抗药物研发就迅速开展起来。利妥昔单抗候选最初是被Ronald Levy医生研究发现,并以此创立了IDEC Pharmaceuticals公司,其研发过程约七年左右。美罗华在美国的市场是由罗氏与Biogen分享。最初,美罗华是以孤儿药的身份被批准进入市场,用于治疗难治B细胞CD20阳性非霍奇金淋巴瘤。相比其他适应症,孤儿药可更加快速地获批进入市场,得到的临床数据有助于美罗华其他适应症的获批。这种以孤儿药身份进入市场,之后拓展至其他适应症的策略被行业广泛效仿。至今,美罗华被批准用于治疗几乎所有的非霍奇金淋巴瘤(Non-Hodgkins Lymphomas)及自身免疫系统疾病等。美罗华自上市后取得了巨大成功,销售额逐年增长,盘踞全球畅销药前十名多年,成为罗氏制药销售前三的抗癌药。然而,生物类似药却对美罗华产生了巨大冲击。2019年11月,美罗华的生物类似药被批准上市,使得美罗华2020年的排名从前10名跌至第17名。拥有基因泰克的罗氏制药作为抗体类药物的领导者,具备强大研发能力。2020年单药销售排名位居第16位的,是另一款基因泰克的产品Ocrevus,为人源化抗CD20单克隆抗体。Ocrevus在2017年3月获FDA批准上市,用于治疗多发性硬化症(Multiple Sclerosis,MS)。Ocrevus上市之后销售强劲,2020年销售额达到64.1亿美元。2.经典案例之二:治疗实体肿瘤、眼科药物新星—抗VEGF抗体药物,Avastin、Lucentis、Eylea基因泰克将阿瓦斯汀(Avastin)推入市场,其有效成分是贝伐珠单抗(Bevacizumab),是全人源的抗VEGF单克隆抗体,通过与血管内皮生长因子(VEGF)结合,抑制肿瘤血管生成,阻断肿瘤供给营养,以达到抗癌的效果。阿瓦斯汀是第一个获批上市的以促血管内皮细胞生长因子为靶点的抗癌药,其适应症包括结直肠癌、非小细胞肺癌等。1989年,基因泰克公司研究员费拉拉(Napoleone Ferrara)和他的同事第一次分离并克隆了VEGF。研究发现VEGF在实体恶性肿瘤细胞往往过度表达,贝伐珠单抗则用于抑制血管生成,产生抗癌效果。2004年,贝伐珠单抗被美国FDA批准作为治疗结直肠癌的药物。其作用机理与当时其他抗癌药物完全不同,是获批上市的第一个抗血管生成的药物。2010年,费拉拉因此发现荣获美国拉斯克临床医学奖(Lasker Award)。阿瓦斯汀多年占据全球单药销售前十,但随着2019年安进与艾尔建推出Mvasi,及2020年初辉瑞制药推出Zirabev,其市场份额被这两款生物类似药抢夺。2020年,阿瓦斯汀位居第14位,销售额为53.2亿美元。值得一提的是,除了对抗肿瘤,抗VEGF药品还可以解决视网膜新生血管问题,治疗老年黄斑变性等眼病。这类疾病在抗VEGF药物出现之前,患者无法逃脱最终失明的结果。贝伐珠单抗的孪生兄弟,雷珠单抗(Ranibizumab)也被基因泰克开发出来,商品名为Lucentis。2012年,美国FDA批准其用于湿性老年黄斑变性,展现出令人欣喜的疗效。然而,专门用于眼科的雷珠单抗的价格极其高昂,而贝伐珠单抗价格远远低于雷珠单抗。并且,美国、中国及欧洲的医生认为其可达到治疗湿性老年黄斑变性的效果,因此在实际临床上会将其作为替代品以Off-label use用于眼病的治疗方案,以减轻患者经济负担。基因泰克从未将贝伐珠单抗适应症拓展到眼科疾病的原因,也引发了各界争议。从技术角度讨论,虽然这两种抗体药同宗同族,但是雷珠单抗的分子量只有贝伐珠单抗的三分之一,用于眼科方面的药物吸收,药物动力学及药物代谢学性质会有很大差别。另外,此类药物的使用方法是眼底注射,对药物的生产要求更高,所以会增加生产成本。从商业角度讨论,拓展适应症本身需要大量的临床试验投入,并且贝伐珠单抗的拓展成功也只能为基因泰克带来雷珠单抗销售的大幅减少。对于制药这个特殊的行业,在竞争日益激烈的环境下维持公司运营发展而追求经济利益,还是牺牲自身利益为患者提供负担得起的药物,是无法避免的伦理悖论。另外一款著名的眼科药物为Eylea,是由美国Regeneron公司开发的融合蛋白抗体类药物。这款药物在2011年被美国FDA批准用于治疗湿性老年黄斑变性,在2014年及2019年又分别获批成为糖尿病黄斑水肿及糖尿病视网膜病变的治疗方法,多年位列美国畅销药排行榜前十。Eylea上市时就展现出了优于Lucentis的特性。首先,体外试验中,Eylea与VEGF的亲和力是雷珠单抗的100倍。其次,在临床上,Lucentis每月进行眼部注射一次,而Eylea则是最初三次注射为每月一次,而后变为只需每两个月一次。再者,Eylea每剂价格要略低于Lucentis,每年使用药物花费只是Lucentis的一半,可大大减轻患者和医疗系统的负担。Eylea上市后销售一直增长,2020年在全球畅销药物中排名第六,全年销售额为83.6亿美元。3.经典案例之三:“伴随诊断”的靶向药物,抗HER2抗体药物,Herceptin,开启精准医疗时代赫赛汀(Herceptin)是需要“伴随诊断”的特异性靶点的抗体药物,1998年被美国FDA批准用于治疗HER2阳性乳腺癌,有效成分为曲妥珠单抗(Trastuzumab),是抗HER2全人源单克隆抗体。1986年,基因泰克的研究员Alex Ullrich发现HER2基因表达出的蛋白,而HER2蛋白可以促使细胞癌变及生长,以此推断HER2蛋白一定与癌症相关,却无法确定适应症。随后,加州大学洛杉矶分校的Dennis Slamon博士发现HER2过度表达的乳腺癌患者恶性程度高,预后差。后来两者通过合作共同开发出曲妥珠单抗。值得注意的是,赫赛汀在1998年9月25日获批,同一天被FDA批准的还有DAKO公司的HER2基因体外检测方法HercepTest。这并不是巧合,而是基因泰克与DAKO公司相辅相成的结果。作用于特定基因型的靶向药物,和为保证药物的有效性而检测患者是否有这种基因型的检测方法同时获批,这开启一个精准治疗的全新时代:联合伴随基因诊断(Companion diagnosis)以预测药物选择和靶标特定基因型药物的诊断治疗方案。HER2阳性乳腺癌患者大约占总乳腺癌患者的25%-30%,赫赛汀在治疗这部分患者上显示了大大超越以往药物的效果。曲妥珠单抗对HER2阳性的乳腺癌病人的突破性疗效使得其很快成为“重磅炸弹”。2010年,其适应症也通过联用化疗药物拓展到HER2阳性胃癌。4.经典案例之四:治疗自身免疫系统疾病,抗TNF-a为抗体药物,Humira雅培公司的单克隆抗体药修乐美(Humira)是最著名的TNF-a抗体药物。修乐美从2012年起至今连续九年蝉联全球最畅销药物,为继立普妥之后的一代药王,2020年销售额高达204亿美元。虽然有多种生物类似药在其2016年及2018年美国及欧洲专利到期依次进入市场,但却从未撼动其销售冠军的地位。修乐美的有效成分是阿达木单抗(Adalimumab),是药物批准时唯一全人源化以TNF-a为靶点的抗体类药物,原理为阻碍TNF受体的启动,抑制与自身免疫系统相关的炎症反应,主要适应症为自身免疫系统类疾病。阿达木单抗于1993年由德国巴斯夫公司(BASF)委托Cambridge Antibody Technology研发。当时这个候选药物称为D2E7,并在1998年时就显现出了良好的临床I期数据。2001年,巴斯夫公司因为公司战略调整,将旗下所有制药业务以仅69亿美元的价格卖给了雅培公司,其中包括D2E7。雅培公司当时因为缺乏强有力的研发管线而被各方诟病,也乐于收购巴斯夫所有的药物管线,并且继续阿达木单抗的研发。这款药物在2002年获得美国FDA批准用于治疗类风湿性关节炎,商品名为修乐美。之后,雅培不断开发新适应症,使得修乐美在市场上更加广泛应用。2012年,雅培公司分拆为两个公司,其中之一就是美国艾伯维公司(AbbVie),拥有修乐美的管理与市场权。一次偶然的收购,为雅培以及后来的艾伯维带来巨大的经济效益。修乐美的成功可以归纳为几个因素:1、更卓越的治疗效果和安全性,医生们更加放心为患者推荐用药;2、雅培公司成功的市场策略。自修乐美上市以来,雅培公司不断投入临床试验,扩展其适应症范围,用于多个自身免疫系统疾病的治疗;3、雅培公司不断开发新的药物剂型和配方,以延长修乐美专利保护期;4、修乐美的价格每年被不断提高。修乐美日趋昂贵为患者及医疗体系带来经济负担,而雅培则声称,价格上涨是由于拓展适应症而开展的临床试验的费用十分高昂,且逐年上涨。在修乐美上市之前,市场上已经有两款以TNF-a为靶点的药物,皆于1998年获批:强生的类客(Remicade),用于治疗克隆氏症(Crohn's disease);及安进和辉瑞的恩博(Enbrel),用于治疗风湿性关节炎。这两种药物虽然销售不如“超级重磅炸弹”修乐美,但同样是“重磅炸弹”药物。直到2018年,由于新型免疫疗法的兴起,才使这两款药物跌出了年度销售前十的席位。2020年,恩博的销售额为63.7亿美元,位居全球单药销售第11名;类客的销售额为41.95亿美元,位居全球单药销售第20名。5.经典案例之五:新型抗炎药物,抗IL-12/23抗体药物,Stelara喜达诺(Stelara)由比利时杨森制药开发,2009年9月获得美国FDA上市批准,用于治疗中度至重度成人斑块型银屑病(Moderate to severe plaquepsoriasis)。其后,喜达诺适应症逐渐扩展,包括:克罗恩氏病(Crohn's disease)、溃疡性结肠炎(Ulcerativecolitis)、银屑病性关节炎(Psoriaticarthritis)等。喜达诺的有效成分为乌斯奴单抗(Ustekinumab),是全人源抗白细胞介素12(IL-12)和白细胞介素23(IL-23)的单克隆抗体。IL-12和IL-23是两种天然存在的细胞因子,被认为可以诱导炎症。而乌斯奴单抗可以与IL-12(p35/p40)与IL-23(p19/p40)所共有的p40亚单位结合,抑制这两种细胞因子,阻止它们与细胞表面的受体IL-12b1结合,阻断下游炎性通路信号传导,进而治疗免疫介导的炎症疾病。喜达诺自上市后,销量由于适应症的拓展及优秀的疗效一直表现不俗。喜达诺的竞争药品类型为JAK抑制剂,FDA于2019年底发布了辉瑞的JAK抑制剂Xeljanz会增加血栓和潜在死亡风险的“黑框警告”,在2021年初,FDA又发布指出对Xeljanz增加心脏问题和癌症的安全担忧。这种状况进一步帮助喜达诺在之后一段时期占领市场。喜达诺在2020年销售额达到79.4亿美元,位居全球单药销售第7名。6.经典案例之六:抗癌免疫疗法(Immunotherapy)的新时代,抗PD-1/PD-L1抗体类药物,Opdivo、Keytruda免疫系统的功能之一是识别自身正常、以及非自身、或者不正常的物质,并对其做出相应反应,从而保证生物机体的正常运作。而其作用机理相当复杂,人类在免疫系统方面的研究已探索数十年,至今也并未厘清所有细节。T细胞是免疫系统工作过程中重要的一环,利用自身受体识别到非自身或者不正常的物质时,就会启动程序清除它们。免疫检查点则是T细胞的“调节器”,表现为T细胞对于一些与自身细胞有差别的物质的容忍度。当T细胞识别到免疫检查点的蛋白时,就会认为这个物质是“好”的,不需要清除。而有些癌细胞就是利用这一点,表达免疫检查点蛋白,而T细胞上相应的免疫检查点蛋白受体与之结合后,T细胞的活动就会被抑制。癌细胞就可以逃过免疫系统的监督,在人体中生长。免疫检查点抑制剂(Immune checkpoint inhibitor)就是“封锁”癌细胞上的免疫检查点蛋白,或者“封锁”T细胞上免疫检查点受体,让两者不能结合,重新启动免疫系统清除癌细胞的力量,利用人体的免疫系统来治疗癌症。来自美国德克萨斯大学安德森癌症研究中心的James P. Allision教授与日本京都大学的本庶佑(Tasuku Honjo)教授发现利用人体自身免疫系统来治疗癌症的机理,并因此在2018年获得了诺贝尔生理学或医学奖。基于此项机理,免疫检查点抑制剂作用于CTLA-4,PD-1/PD-L1的抗体类药物已经获批上市,开启免疫疗法的新纪元,创造出“超级重磅炸弹”药物,进入全球单药销售前十名。这些药物对于癌细胞的作用是间接的,是通过调节T细胞的抗肿瘤免疫应答而发挥作用。与之前介绍的抗癌药物机理非常不同。第一个被批准上市的免疫检查点抑制剂单抗是全人源化抗CTLA-4抗体易普利姆玛(Ipilimumab),商品名为益伏(Yervoy),是由百时美施贵宝(Bristol Myers Squibb)在2011年3月推入市场的。CTLA-4是一种在T细胞表面表达的免疫检查点受体,易普利姆玛与之结合,就可以启动T细胞识别那些表达CTLA-4受体的癌细胞的能力。美国FDA批准益伏用于治疗不可切除或转移性成人恶性黑色素瘤。关于CTLA-4的研究始于获得诺贝尔奖的James P. Allision教授。针对CTLA-4的药物易普利姆玛的研究则在1995年被授权给了一家美国小公司NeXstar Pharmacueticals,随后不久这家公司便被美国吉利德公司(Gilead Sciences)收购。之后,这条研发管线又被吉利德公司在1999年授权给了美国Medarex公司。百时美施贵宝在2009年以24亿美元的价格收购Medarex,就是看上了易普利姆玛这条已经在临床阶段的研发管线。百时美施贵宝大量投入促进其发展,直至推入市场。虽然益伏为第一个批准上市的免疫检查点抑制剂,但副作用比较大,之后销售量一直不敌以PD-1为靶点的可瑞达(Keytruda,也称K-药)和欧狄沃(Opdivo,也称O-药)。PD-1是另一种在T细胞表面表达的免疫检查点受体,将其抑制,则可以让T细胞开始识别可以表达PD-1的受体:PD-L1或者PD-L2蛋白的癌细胞。K-药的有效成分是帕博利珠单抗(Pembrolizumab),是全人源化PD-1抗体,由默沙东公司(Merck Sharp & Dohme)推入市场,在2014年9月被美国FDA经“加速批准”通道,作为孤儿药批准用于治疗不可切除或转移性成人恶性黑色素瘤。O-药是由百时美施贵宝推入市场,其有效成分为纳武利尤单抗(Nivolumab),是全人源化PD-1抗体,在2014年12月获批,同样作为孤儿药批准用于治疗不可切除或转移性成人恶性黑色素瘤。获批之后,两家公司都积极拓展自家药物的适应症,并且大力投入开展各种联合疗法的临床试验。直至今日,这两款药物的适应症都已十分广泛。这两款药物机理相似,所以有些适应症有重叠。而默沙东对于拓展K-药的适应症极为激进,现在适应症涵盖数十种癌症,而包含K-药的联合疗法已经跻身于几个适应症的一线疗法。O-药的适应症开发稍微落后于K-药。自2018年开始,这两款药物双双跻身全球药物单药销售的前十。2020年最畅销药物中,K-药及O-药分别排名第二及第八,年度销售额为177亿美元及79亿美元。K-药和O-药也是制药行业的传奇。之前提及,百时美施贵宝是看中了易普利姆玛而收购Medarex。Medarex除此之外还有其他的研发管线,O-药就是其中之一。因为O-药当时还处在研发的早期阶段,所以在这场并购中并没有引起各方的注意。百时美施贵宝在收购了Medarex之后依然投入研发其他管线,所以使得以PD-1为靶点的O-药脱颖而出,并且在临床阶段展现出了喜人的效果。百时美施贵宝非常幸运地以并不算昂贵的价格收购一家公司,得到了两个靶点不同的优秀免疫检查点抑制剂候选药物,而O-药甚至显示了优于易普利姆玛的效果。百时美施贵宝将这条管线候选药物的临床试验命名为“CheckMate”(将军)系列,足见其寄予厚望。2010年,百时美施贵宝在《新英格兰医学杂志》上发表文章,阐述免疫检查点抑制剂是抗癌药物的强有力候选。而行业巨头默沙东公司(Merck Sharp & Dohme)看到这个数据,预感到以PD-1为靶点的单抗的前途,仔细查看项目储备,惊喜地发现也有一款以PD-1为靶点的单抗研发管线帕博利珠单抗,并集中大量资源专攻这条管线。当时,默沙东的做法存在很大的风险:1、百时美施贵宝的O-药研发进度远远快于默沙东的K-药,有可能优先上市且效果更优。2010年,百时美施贵宝已有临床数据,而默沙东在年底才拿到IND;2、默沙东当时原本就苦于没有强有力的药物管线,如果这场博弈失败,不仅会占用其他在研管线发展所需要的资源,还会进一步加大资金压力。然而,制药史的发展历程总是会让人意想不到:默沙东的K-药早于百时美施贵宝的O-药几个月优先获得了美国FDA的上市批准。之后,K-药在拓展适应症上也处处领先。而默沙东后来者居上,且处处占有先机的重要原因之一,就是对药物机理的判断与临床试验的设计。在开发的过程中,有两种选择:第一种选择,就是将候选药物与一种伴随诊断的方法联合,将药物的使用局限在某一适应症且有相应生物标志物(Biomarker)表达的患者上。对PD-1药物而言,就是看患者癌细胞是否表达PD-L1的基因或者蛋白。这种临床设计思路,可以减少符合入选临床试验的病人数目,进而缩短临床试验所需时间,并且因为有生物标记物表达作为指导,有潜力提高入选患者对药物的应答率以及疗效,进而提高临床试验成功的几率。然而,这样做从经验上看,却会大大限制药物上市以后的适用范围,因为药物上市后也只能用于表达生物标记物的患者,严重影响药物的销售。对于研发费用极其昂贵的制药行业,这种情况是药厂极力要避免的;而另一种选择,就是不考虑伴随诊断方法,直接研究开发药物针对某一适应症。为了赶超百时美施贵宝的研发进度,默沙东选择了第一种方法,而百时美施贵宝则选用了第二种。而就是这个PD-L1生物标记物的检测数据,明确彰显了抗PD-1单抗在此患者群体中的突出疗效。也正因为默沙东收集了这个数据,使得在拓展适应症上领先。而这个事实预示了精准治疗药物是未来行业发展的方向。另一个值得一提的免疫检查点抑制剂单抗则为阿替利珠单抗(Atezolizumab),商品名为泰圣奇(Tecentriq),是由基因泰克在2016年推入市场的,被FDA批准治疗膀胱癌。这是美国FDA批准的第一个抗PD-L1的单抗药物,同样需要PD-L1生物标记物的伴随检测。基因泰克将这款药定位为治疗实体瘤的疗法,其适应症也拓展到非小细胞肺癌、小细胞肺癌、三阴乳腺癌和肝癌等。基因泰克也拓展阿替利珠单抗各种联合疗法。泰圣奇在市场上表现亦是不俗,在2020年销售达到约27亿瑞士法郎。1.传统的全球制药行业领导者通过并购的方式进入生物制药领域,构建核心竞争能力,例如:罗氏制药收购基因泰克、辉瑞制药收购惠氏制药、雅培制药收购德国巴斯夫制药业务。2.生物制药领域的突破性创新,或者临床里程碑式的药品,一般不是被战略规划出来的结果,例如:PD-1的O-药及K-药。正如罗氏制药的CEO Severin Schwan所说:“科学的成功不能被规划,但我们可以创造条件,使之得以实现;我们需要对新想法持开放态度,勇于冒险,并偶尔挑战大众普遍持有的观点;我们的研究人员需要自由地研究他们的想法,给他们充足的时间和持久努力的支援”。
3.以“孤儿药身份”认证为开始,一些突破性创新药品逐步扩大适应症或者专注在一些治疗领域,成为“重磅炸弹”,有的产品甚至全球年销售额超过100亿美元。4.生物制药公司在成长过程中,基本上都面临资金缺乏的困境。专利及经验积累往往是公司最大的资产。
注:本文节选自由香港商务印书馆出版的《生命科技投资启示录·捕捉下一只独角兽》,文章已获出版社及作者授权。(欢迎转发,请联系:shuisongye)
柳达先生是一位药剂师,但更是一位投资者,双重身份的结合令他眼光独到。独角兽是科学家的梦想,也是投资者的追求。
严正声明
本文旨在知识共享,无任何商业用途,如涉及侵权,请及时联系我们。










请先 登录后发表评论 ~